China CDC shares latest COVID-19 data
The Chinese Center for Disease Control and Prevention shared via GISAID new hCoV-19 genome sequences from 27 provinces, collected between 2022-01-01 and 2022-03-30.
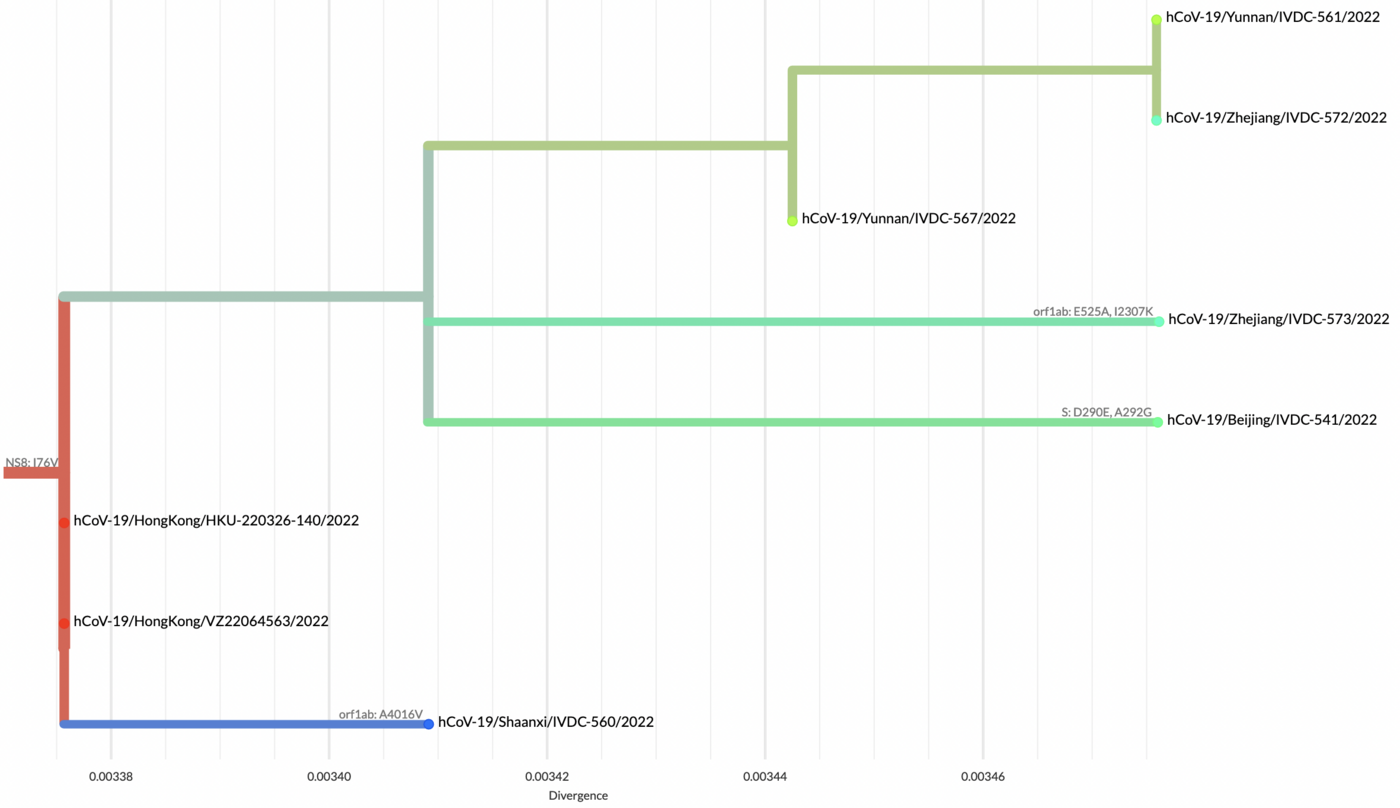
Phylogenetic analyses suggest at least 35 independent introductions from outside China rather than extensive local transmission. This indicates that measures are mostly effective in disrupting long transmission chains following import.
Eleven genomes were assigned as Delta (from six distinct lineages), and 66 genomes were assigned as Omicron (from 14 distinct lineages). No highly divergent new variants were found.
The full map including putative transmission links from the genomic tree distance is shown here:
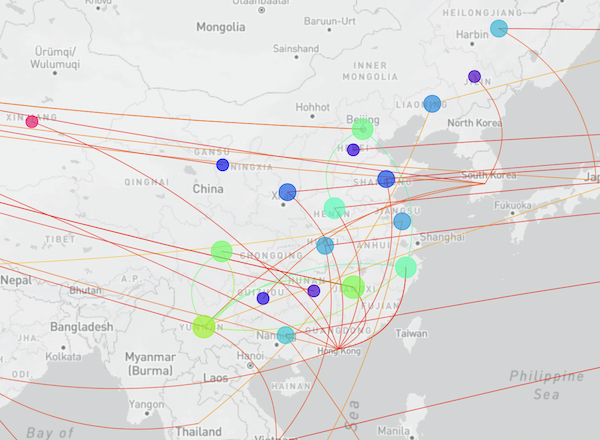
Searching the 77 new hCoV-19 genome sequences with GISAID’s AudacityInstant against the 10.6 million genomes in the EpiCoV database found a total of 16,278 closely related genomes. The graphic below shows the distribution of countries of the closest hits of which the dark blue bars correspond to identical sequence hits from abroad and possible transmission links:
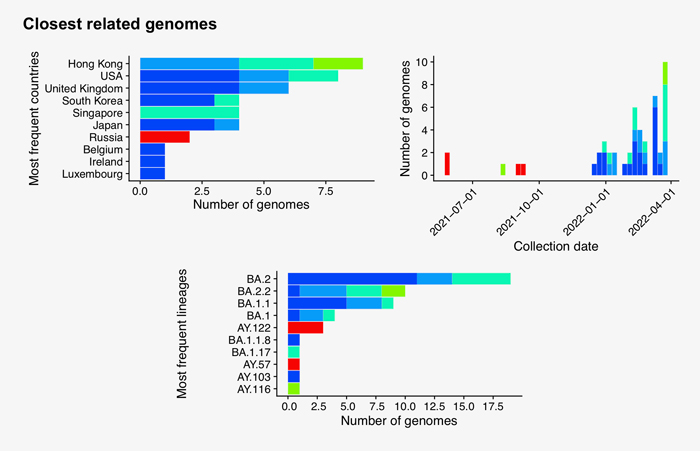
Full phylogenetic analysis with the latest shared hCoV-19 genome sequences from China shown as large circles together with nearest hits from EpiCoV (identified with GISAID’s AudacityInstant) illustrates the pattern of individual introductions. The second panel shows the detailed lineages colored in the same tree.
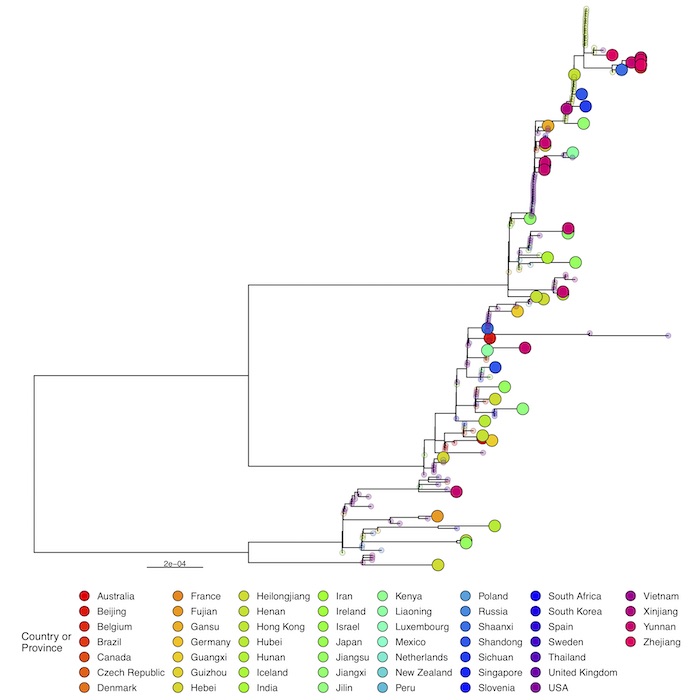
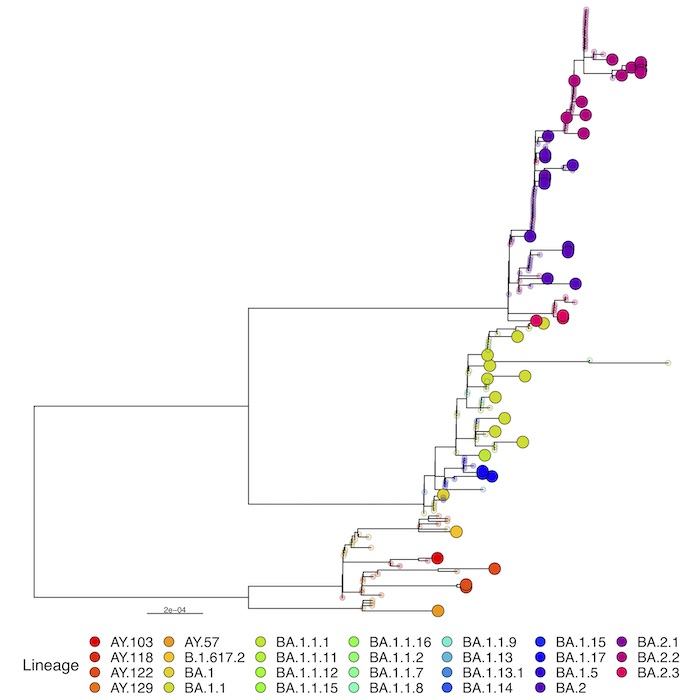
A small cluster of multiple cities is seen in the most divergent tip with Hong Kong cases in the genetic base which are part of lineage BA.2.2 which is more common in Asia. The detail of this cluster is below (click image to enlarge):